. DOI: 10.1186/s13321-023-00769-x")
Biomedical engineers at Duke University have developed an AI platform that autonomously compares molecules and learns from their variations to anticipate property differences critical to discovering new pharmaceuticals. The platform provides researchers with a more accurate and efficient tool to help design therapeutics and other chemicals with useful properties.
The research was published on October 27 in the Journal of Cheminformatics.
Machine learning algorithms are increasingly used to study and predict the biological, chemical and physical properties of small molecules used in drug development and other material design tasks. These tools can help researchers understand the key “ADMET” properties of a molecule—how it’s absorbed, distributed, metabolized, excreted and its toxicity within the body. By understanding these different properties, researchers can identify molecules to develop new therapeutics that are safer and more effective.
While existing machine learning platforms enable researchers to screen a much larger number of molecules than would be possible by physically making them all in a lab, they can only predict the properties of one molecule at a time, limiting their overall efficiency when tasked to identify the most optimal compound.
While there are a few other computational approaches to cut out this extra step and directly compare molecules, they are limited in their scope. For example, methods such as free energy perturbation are very accurate but so computationally complex that they can only evaluate a handful of molecules at a time. Approaches such as matched molecular pairs, on the other hand, are much faster but can only compare very similar molecules, limiting their wider use.
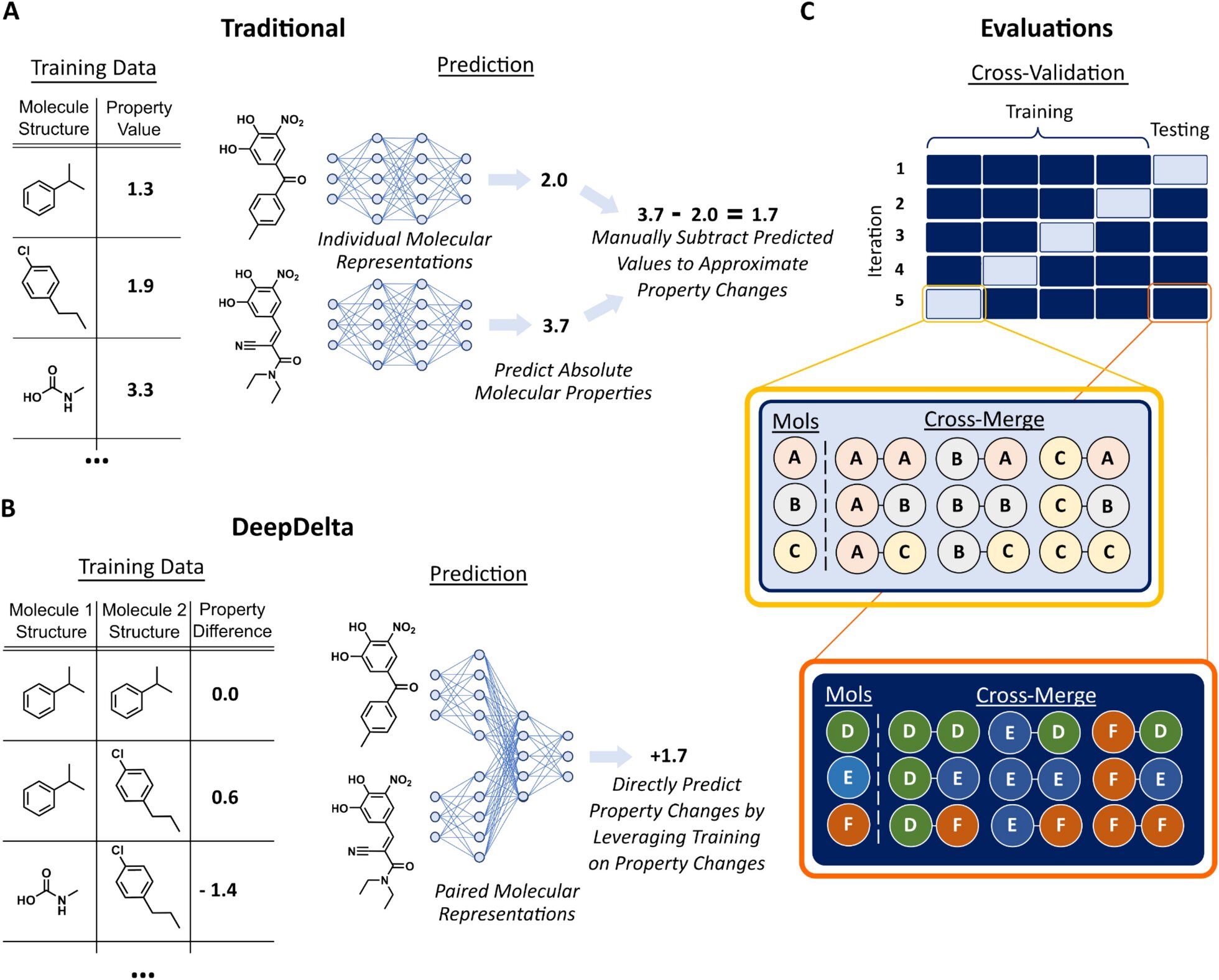
To address this issue, Reker and Zachary Fralish, a Ph.D. student in the Reker lab, developed DeepDelta, a deep learning approach that can efficiently compare two molecules simultaneously and predict the property differences between them even if they are very different.
“By having the network learn from a one-to-one comparison, you’re giving it more data points than if it was learning from one molecule at a time,” said Reker. “The platform is learning about the structure and properties of each molecule individually, but it’s also learning about the differences between the two and how those differences inform the molecule’s properties.”
The team tested the DeepDelta platform against two state-of-the-art models in the field: Random Forest, a widely used classic machine learning model, and ChemProp, a deep neural network that DeepDelta is based off of. Each system compared two known molecular structures and predicted 10 different ADMET properties, including how the molecules are cleared from the kidneys, their respective half-lives and how well they can be metabolized by the liver.
DeepDelta proved significantly more effective and precise at predicting and quantifying the differences in molecular properties between molecules than the existing platforms.
“Training on molecular differences allows this method to be more accurate when deciding if a new chemical is better or worse than a current one,” said Fralish. “It’s like doing homework that is more like your test. We also greatly expanded the size of our datasets by pairing, essentially giving our models more homework, which really helps data-hungry neural networks learn more.”
The team is now looking forward to incorporating this model into their work as they design potential new therapies and optimize existing drug candidates.
“With this tool, we could look at a drug that almost made it through FDA approval, but maybe it had issues with liver toxicity, so it didn’t quite make it,” said Fralish. “DeepDelta could help identify molecules that have those same good properties but no liver toxicity. This tool opens up a lot of opportunities by helping us decide what chemical has the best chance of doing what we want in the real world, saving time and money.”
More information:
Zachary Fralish et al, DeepDelta: predicting ADMET improvements of molecular derivatives with deep learning, Journal of Cheminformatics (2023). DOI: 10.1186/s13321-023-00769-x
Provided by
Duke University
Citation:
AI model directly compares properties of potential new drugs (2023, December 4)
retrieved 4 December 2023
from https://phys.org/news/2023-12-ai-properties-potential-drugs.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no
part may be reproduced without the written permission. The content is provided for information purposes only.





